The HIRUZ Clinical Trial Unit (CTU) is recognized for its expertise in the support and advice in submissions, project management and monitoring of academic clinical research projects. Investigator-initiated academic clinical research is defined as research for which a (university) hospital, university or an authorized organization acts as sponsor. This means that this particular institution has the full responsibility over the design, set-up, management and safety of the clinical experiment.
In addition, the sponsor owns the study results and has the right to publish them freely. The definition of investigator-initiated research does not exclude that the institution receives support in order to execute the study (e.g. financial support, free medication, statistical support, CRF creation, personnel, …).
Tom Verschoore | Clinical Trial Unit Manager
Standard services for studies performed by Ghent University (Hospital)
- Support in the preparation of a correct and complete submission package for the Ethics Committee and Competent Authority, if applicable
- Guiding the investigator in reporting to the Ethics Committee and/or the Competent Authority: amendments, safety reporting, annual progress report, end of study reporting, notifications,…
- Performing on-site and remote monitoring according ICH-GCP for all clinical trials with medical products and medical devices for which Ghent University (Hospital) is sponsor. Monitoring can also be provided for other trials upon request,
- Providing a no-fault insurance (only when Ghent University (Hospital) is sponsor),
- Guiding investigators and providing advice in the development of a trial,
- Providing (online) training regarding ICH-GCP
- Follow-up on the new or updated regulations, interact with other research institutions (universities, university colleges, VIB, Government, KCE, …)
Contact
Health, innovation and research institute
Clinical Trial Unit
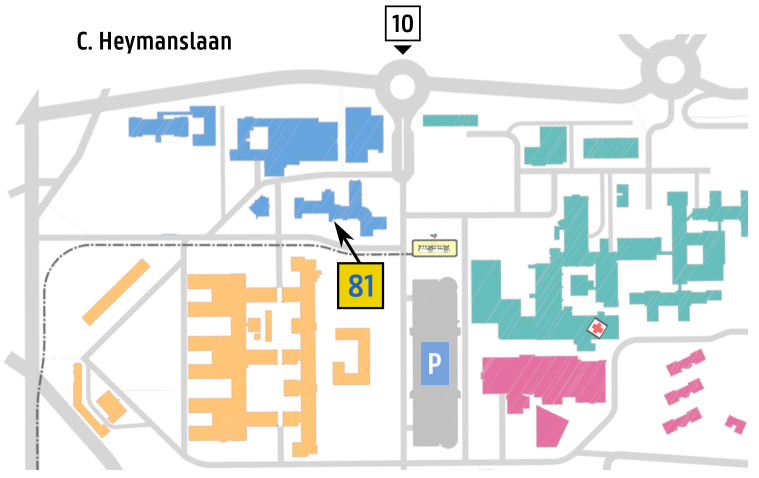
Entrance 81 / Route 812
C. Heymanslaan 10, 9000 Gent
Before setting-your trial
hiruz.ctu@uzgent.be
+32 9 332 05 30
Study related questions
Contact your focus team